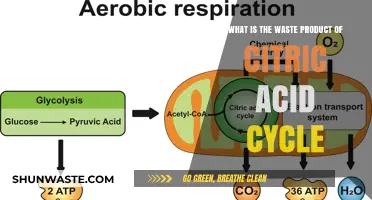
Amino acid metabolism is a crucial biological process that involves the breakdown and synthesis of amino acids, which are the building blocks of proteins. As these essential molecules are metabolized, they produce various by-products, with one of the primary waste products being ammonia (NH3). This highly toxic substance is generated when the amino group (-NH2) is removed from amino acids during catabolic reactions. The body has evolved efficient mechanisms to handle this waste, primarily through the urea cycle in the liver, where ammonia is converted into urea, a less toxic compound that can be safely excreted in urine. Understanding the waste products of amino acid metabolism is vital in fields like biochemistry and physiology, as it provides insights into nutrient utilization, waste management, and the potential health implications of metabolic disorders.
| Characteristics | Values |
|---|---|
| Primary Waste Product | Ammonia (NH₃) |
| Form of Excretion | Urea (in most mammals, including humans) |
| Process of Conversion | Urea Cycle (Ornithine Cycle) |
| Organs Involved in Conversion | Liver (primary site of urea synthesis) |
| Other Waste Products | Uric acid (in birds, reptiles, and some insects) |
| Toxicity of Primary Waste | Ammonia is highly toxic to cells; conversion to urea or uric acid reduces toxicity |
| Energy Requirement | Urea synthesis is energy-intensive, requiring ATP |
| Excretion Route | Kidneys (urea is excreted in urine) |
| Alternative Pathways | Transaminations, deamination, and oxidative deamination |
| Clinical Significance | Elevated ammonia levels can lead to hepatic encephalopathy; urea cycle disorders are genetic conditions affecting waste removal |
Explore related products






What You'll Learn
- Urea Cycle: Process converting toxic ammonia from amino acid breakdown into urea for safe excretion
- Ammonia Production: Amino acid deamination releases ammonia, a waste product requiring detoxification
- Ketone Bodies: Formed from amino acid breakdown, ketones are alternative energy sources during fasting
- Creatine Metabolism: Creatine breakdown produces creatinine, excreted as waste via kidneys
- Sulfur-Containing Amino Acids: Metabolism of methionine and cysteine produces sulfur waste, excreted as sulfate
![]()
Urea Cycle: Process converting toxic ammonia from amino acid breakdown into urea for safe excretion
Amino acid metabolism is a vital process that supports protein synthesis, energy production, and cellular repair. However, it also generates a toxic byproduct: ammonia. Left unchecked, ammonia accumulates in the bloodstream, disrupting pH balance and damaging the brain and other organs. The urea cycle, a metabolic pathway primarily occurring in the liver, mitigates this threat by converting ammonia into urea, a far less toxic compound safely excreted in urine.
Understanding this cycle is crucial for appreciating the body's intricate waste management system and for identifying disorders that disrupt it, such as urea cycle disorders, which can lead to severe neurological complications if untreated.
The urea cycle involves a series of enzymatic reactions that combine ammonia with carbon dioxide to form urea. It begins with the conversion of ammonia to carbamoyl phosphate, catalyzed by the enzyme carbamoyl phosphate synthetase I (CPS1). This step requires the presence of ornithine, a non-protein amino acid, and consumes one molecule of ATP. The carbamoyl phosphate then reacts with ornithine to form citrulline, facilitated by ornithine transcarbamylase (OTC). Citrulline is transported to the cytoplasm, where it combines with aspartate to form argininosuccinate, a reaction catalyzed by argininosuccinate synthetase (ASS). Finally, argininosuccinate lyase (ASL) cleaves argininosuccinate into arginine and fumarate. Arginine is then hydrolyzed by arginase to produce urea and regenerate ornithine, completing the cycle. Each turn of the cycle consumes three molecules of ATP and one molecule of ammonia, highlighting its energy-intensive nature.
Disruptions in the urea cycle can have severe consequences, particularly in infants and young children. Urea cycle disorders (UCDs) are a group of genetic conditions where one of the enzymes involved in the cycle is deficient or absent. For example, ornithine transcarbamylase deficiency, the most common UCD, leads to a buildup of ammonia because the conversion of carbamoyl phosphate to citrulline is impaired. Symptoms of UCDs include poor feeding, vomiting, lethargy, and seizures, often manifesting within the first few days of life. Early diagnosis through newborn screening and prompt treatment with medications like sodium benzoate and arginine, along with a low-protein diet, are critical to managing these disorders and preventing irreversible brain damage.
For individuals with UCDs, dietary management is a cornerstone of treatment. Protein intake must be carefully monitored, as excessive amino acid breakdown can overwhelm the compromised urea cycle. Infants with UCDs may require specialized formulas that are low in protein but supplemented with essential amino acids. Older children and adults often need to limit high-protein foods like meat, dairy, and legumes, while ensuring adequate caloric intake from carbohydrates and fats. Regular monitoring of blood ammonia levels and periodic consultations with metabolic specialists are essential to adjust treatment plans and prevent metabolic crises.
In summary, the urea cycle is a metabolic safeguard that transforms toxic ammonia into urea, enabling its safe elimination from the body. Its efficiency is critical for health, particularly in vulnerable populations like newborns. Understanding this process not only sheds light on the body's waste management mechanisms but also underscores the importance of early detection and management of disorders that disrupt it. By balancing dietary protein intake and leveraging medical interventions, individuals with urea cycle disorders can lead healthier lives, minimizing the risk of ammonia toxicity and its devastating consequences.
Unlocking Hidden Potential: The Surprising Value of Wasted Resources
You may want to see also
Explore related products






![]()
Ammonia Production: Amino acid deamination releases ammonia, a waste product requiring detoxification
Amino acid metabolism is a fundamental process in the human body, essential for protein synthesis, energy production, and cellular repair. However, this vital process also generates waste products that must be managed to prevent toxicity. One such waste product is ammonia, a highly toxic compound that is released during the deamination of amino acids. This process, while necessary for breaking down amino acids into usable components, poses a significant challenge due to ammonia's potential to cause severe harm if not promptly detoxified.
The deamination of amino acids occurs primarily in the liver, where enzymes such as glutamate dehydrogenase and transaminases catalyze the removal of an amino group (-NH₂) from the amino acid molecule. This amino group is then converted into ammonia (NH₃). While ammonia is a natural byproduct of protein metabolism, its accumulation can lead to hyperammonemia, a condition characterized by elevated blood ammonia levels. In healthy individuals, the body efficiently detoxifies ammonia through the urea cycle, a series of biochemical reactions that convert ammonia into urea, a less toxic substance that is excreted in urine. However, disruptions in this cycle, whether due to genetic disorders, liver disease, or certain medications, can result in dangerous ammonia buildup.
Detoxification of ammonia is particularly critical in vulnerable populations, such as infants and individuals with liver dysfunction. For example, newborns, especially preterm infants, have an immature urea cycle, making them more susceptible to hyperammonemia. Similarly, patients with liver diseases like cirrhosis or hepatitis often experience impaired ammonia detoxification, leading to complications such as hepatic encephalopathy, a condition where ammonia affects brain function. To mitigate these risks, medical interventions such as sodium benzoate or sodium phenylbutyrate may be administered. These medications act as alternative pathways for ammonia detoxification, conjugating with glycine to form urinary excretable products, thereby reducing ammonia levels in the blood.
Understanding the role of ammonia in amino acid metabolism also highlights the importance of dietary considerations. High-protein diets, while beneficial for muscle growth and repair, can increase the burden on the body's detoxification systems by producing more ammonia. Individuals with compromised liver function or genetic disorders affecting the urea cycle should monitor their protein intake and consult healthcare providers for personalized dietary recommendations. For instance, a low-protein diet supplemented with essential amino acids may be advised to minimize ammonia production while ensuring adequate nutrition.
In conclusion, ammonia production from amino acid deamination is a double-edged sword—essential for nutrient utilization yet hazardous if not properly managed. The body's detoxification mechanisms, particularly the urea cycle, play a pivotal role in maintaining ammonia homeostasis. However, when these systems fail, the consequences can be life-threatening. Awareness of risk factors, early detection of hyperammonemia, and targeted interventions are crucial for preventing ammonia-related complications. By balancing metabolic needs with effective detoxification strategies, individuals can safeguard their health while reaping the benefits of amino acid metabolism.
Weathering, Erosion, and Mass Wasting: Understanding Earth's Surface Processes
You may want to see also
Explore related products

$23.1 $24.78





![]()
Ketone Bodies: Formed from amino acid breakdown, ketones are alternative energy sources during fasting
Amino acid metabolism, a complex process vital for energy production and cellular function, generates various byproducts, including ketone bodies. These compounds, often associated with low-carb diets and fasting, are a fascinating example of the body's adaptability in energy management. When carbohydrates are scarce, the body turns to alternative fuel sources, and this is where ketones step in, offering a unique metabolic pathway.
The Ketogenic Shift: During prolonged fasting or strict carbohydrate restriction, the body's preferred energy source, glucose, becomes limited. In response, the liver initiates a process called ketogenesis, breaking down fatty acids and, notably, amino acids. This metabolic shift is a survival mechanism, ensuring energy supply to vital organs, especially the brain, which typically relies on glucose. Ketone bodies, including acetoacetate, beta-hydroxybutyrate, and acetone, are the result of this metabolic detour.
Amino Acids' Role: Here's where amino acid metabolism's waste product comes into play. When amino acids are broken down, they can follow different pathways. Some amino acids, particularly those with a carbon skeleton that can be easily converted, contribute to ketone body formation. This process is particularly active during fasting when the body prioritizes energy extraction from various sources. For instance, the amino acid leucine can be converted into acetyl-CoA, a precursor to ketone bodies, providing an alternative energy substrate.
Practical Implications: Understanding this metabolic flexibility has led to the popularity of ketogenic diets, which aim to induce a state of ketosis. In this state, the body primarily uses ketones for energy. For individuals considering such diets, it's essential to note that the transition period can be challenging. Initially, as the body adapts, one might experience the 'keto flu,' characterized by fatigue and brain fog. However, once adapted, many report increased energy levels and improved mental clarity. It's crucial to ensure adequate protein intake to preserve muscle mass, as excessive amino acid breakdown for energy can lead to muscle wasting.
Fasting and Ketones: Intermittent fasting, a practice gaining traction, also leverages this metabolic pathway. During fasting periods, typically lasting 16-24 hours, the body gradually shifts from glucose to ketone bodies for energy. This transition is a natural process, and for most healthy individuals, it is safe and can offer various health benefits. However, it's imperative to stay hydrated and maintain electrolyte balance during fasting, as the body's fluid requirements may change. For those new to fasting, starting with shorter durations and gradually increasing is advisable.
In summary, ketone bodies, formed partly from amino acid breakdown, are a testament to the body's metabolic versatility. This process, while complex, can be harnessed through dietary and lifestyle choices, offering an alternative energy source during fasting or specific dietary regimens. As with any metabolic intervention, understanding the underlying biology is key to optimizing its benefits while mitigating potential risks.
Understanding the Alarming Percentage of Paper Waste Globally
You may want to see also
Explore related products






![]()
Creatine Metabolism: Creatine breakdown produces creatinine, excreted as waste via kidneys
Creatine, a compound synthesized from amino acids like arginine, glycine, and methionine, plays a pivotal role in energy production within muscle cells. However, its metabolism inevitably leads to the formation of creatinine, a waste product that serves as a biomarker for kidney function. This process begins when creatine phosphate donates a phosphate group to regenerate ATP, the cell’s energy currency, during high-intensity activities. Over time, creatine undergoes spontaneous degradation, converting into creatinine at a rate of approximately 1-2% daily. This breakdown is irreversible, making creatinine a natural byproduct of creatine turnover in the body.
The excretion of creatinine is a critical aspect of amino acid metabolism, primarily handled by the kidneys. Unlike creatine, which is actively reabsorbed in the kidneys, creatinine is freely filtered and excreted in urine. Healthy individuals typically excrete 1-2 grams of creatinine daily, depending on muscle mass and dietary intake. Elevated levels in blood or urine can signal impaired kidney function, as the organs struggle to clear this waste product efficiently. Monitoring creatinine levels is thus a standard diagnostic tool for assessing renal health, particularly in athletes or individuals supplementing with creatine.
For those considering creatine supplementation, understanding its metabolic pathway is essential. A common dosage of 3-5 grams daily can enhance muscle performance by increasing intracellular creatine stores. However, this also elevates creatinine production proportionally. While this is generally harmless for individuals with healthy kidneys, those with pre-existing renal conditions should exercise caution. Staying hydrated and avoiding excessive protein intake can support kidney function, ensuring efficient creatinine clearance. Regular blood tests to monitor creatinine levels are advisable for long-term users.
Comparatively, creatine metabolism contrasts with other amino acid pathways, where waste products like urea are produced in the liver and excreted via urine. Creatinine’s direct renal excretion makes it a unique marker of both muscle metabolism and kidney efficiency. Unlike urea, which fluctuates with protein intake, creatinine levels are relatively stable, reflecting consistent creatine breakdown. This stability makes it a reliable indicator of renal health, though it underscores the importance of kidney function in managing amino acid waste products.
In practical terms, individuals can optimize creatine metabolism by balancing supplementation with adequate hydration and a moderate protein diet. For older adults or those with reduced kidney function, lower doses (2-3 grams daily) may be safer. Pairing creatine with carbohydrates can enhance absorption and minimize stress on metabolic pathways. Ultimately, while creatinine is a natural waste product, its management through mindful supplementation and renal care ensures both performance benefits and long-term health.
Understanding Standard RV Waste Container Sizes for Efficient Travel
You may want to see also
Explore related products


$19.89 $21.99



![]()
Sulfur-Containing Amino Acids: Metabolism of methionine and cysteine produces sulfur waste, excreted as sulfate
Amino acid metabolism is a complex process that not only fuels protein synthesis but also generates waste products that the body must eliminate. Among the various amino acids, sulfur-containing amino acids like methionine and cysteine play a unique role due to their sulfur content. When these amino acids are metabolized, the sulfur they contain is not stored in the body but must be processed and excreted. This sulfur waste is primarily eliminated as sulfate, a process that involves multiple organs and biochemical pathways.
The metabolism of methionine, an essential amino acid, begins with its conversion to S-adenosylmethionine (SAM), a critical molecule in methylation reactions. As SAM donates methyl groups, it is converted to S-adenosylhomocysteine (SAH), which is then hydrolyzed to homocysteine. Homocysteine can either be remethylated back to methionine or enter the transsulfuration pathway, where it is converted to cysteine. Cysteine, another sulfur-containing amino acid, can also be derived directly from dietary sources or synthesized from methionine. Both pathways ultimately produce sulfate as a byproduct, which is then excreted in urine. This process highlights the body’s efficient system for managing sulfur, ensuring it does not accumulate to toxic levels.
From a practical standpoint, understanding sulfur waste from amino acid metabolism is particularly relevant for individuals with specific dietary or health conditions. For example, high intake of methionine-rich foods like red meat, eggs, and dairy can increase sulfur waste production, potentially straining the kidneys. Conversely, vegetarians or vegans may consume less methionine but more cysteine from plant sources like legumes and nuts. Monitoring sulfur intake and ensuring adequate hydration can support the kidneys in efficiently excreting sulfate. For older adults or those with renal impairment, reducing methionine intake and increasing antioxidant-rich foods may help mitigate oxidative stress associated with sulfur metabolism.
Comparatively, sulfur metabolism differs from other amino acid waste products like urea, which is produced from the breakdown of non-sulfur amino acids. While urea is primarily excreted through urine, sulfate excretion involves additional steps, including oxidation in the liver and kidneys. This distinction underscores the importance of sulfur-specific metabolic pathways and their regulation. For instance, the enzyme sulfite oxidase plays a critical role in converting sulfite to sulfate, a reaction essential for preventing sulfite toxicity. Genetic deficiencies in this enzyme can lead to severe metabolic disorders, emphasizing the delicate balance required in sulfur metabolism.
In conclusion, the metabolism of sulfur-containing amino acids like methionine and cysteine is a finely tuned process that generates sulfate as a waste product. This sulfate is efficiently excreted through urine, supported by organs like the liver and kidneys. Practical considerations, such as dietary intake and hydration, can influence this process, particularly in vulnerable populations. By understanding these mechanisms, individuals can make informed choices to support their metabolic health and ensure the safe elimination of sulfur waste.
Recycling Silver Waste: Analytical Chemistry Methods for Sustainable Recovery
You may want to see also
Frequently asked questions
The primary waste product of amino acid metabolism is urea, which is produced in the liver through the urea cycle and excreted by the kidneys.
Urea is formed through the urea cycle, where ammonia (a toxic byproduct of amino acid breakdown) is converted into urea. This process involves enzymes like carbamoyl phosphate synthetase and arginase.
Yes, besides urea, amino acid metabolism also produces ammonia and water. Ammonia is converted to urea to prevent toxicity, while water is a byproduct of deamination reactions.








![SMARTERNUTRITION Essential Amino Acids (EAA) Capsules - 1 Gram Per Serving of All 9 EAAs - Supports Muscle Mass & Exercise - Non-GMO, Vegan, Gluten Free - 60 Count[30-Day Supply]](https://m.media-amazon.com/images/I/718lHgi4dgL._AC_UL320_.jpg)