21-hydroxylase deficiency, a common form of congenital adrenal hyperplasia (CAH), is primarily classified into two major phenotypes based on its clinical presentation: simple-virilizing and salt-wasting. The distinction between these forms hinges on the severity of the enzyme deficiency and its impact on adrenal hormone production. In simple-virilizing CAH, the deficiency is partial, allowing for some cortisol and aldosterone synthesis, which prevents life-threatening salt-wasting crises but leads to excessive androgen production, causing prenatal virilization of female fetuses. Conversely, the salt-wasting form results from a more severe enzyme deficiency, leading to profound cortisol and aldosterone deficiency, which manifests as hyponatremia, hyperkalemia, and dehydration, often requiring immediate medical intervention. Understanding the differences between these phenotypes is crucial for accurate diagnosis, management, and long-term outcomes in affected individuals.
| Characteristics | Values |
|---|---|
| Type of 21-Hydroxylase Deficiency | Can be either Simple-Virilizing or Salt-Wasting, depending on severity |
| Enzyme Deficiency | Deficiency of 21-hydroxylase enzyme (CYP21A2) |
| Affected Hormones | Cortisol, aldosterone, and androgens |
| Simple-Virilizing Form | - Partial enzyme deficiency - Mild cortisol deficiency - Normal aldosterone production - Virilization in females (e.g., ambiguous genitalia) |
| Salt-Wasting Form | - Severe enzyme deficiency - Severe cortisol and aldosterone deficiency - Life-threatening salt-wasting crisis (e.g., hyponatremia, hyperkalemia) - Virilization in females |
| Clinical Presentation (Simple-Virilizing) | - Ambiguous genitalia in females - No salt-wasting crisis - Normal blood pressure and electrolytes |
| Clinical Presentation (Salt-Wasting) | - Ambiguous genitalia in females - Salt-wasting crisis in neonates - Dehydration, shock, and electrolyte imbalances |
| Diagnosis | - Newborn screening (elevated 17-OHP) - Hormonal assays (cortisol, aldosterone, ACTH) - Genetic testing (CYP21A2 mutations) |
| Treatment (Simple-Virilizing) | - Glucocorticoid replacement (e.g., hydrocortisone) - Monitoring for adrenal insufficiency |
| Treatment (Salt-Wasting) | - Glucocorticoid and mineralocorticoid replacement (e.g., hydrocortisone + fludrocortisone) - Emergency management of salt-wasting crises |
| Prognosis | - Good with early diagnosis and treatment - Risk of adrenal crisis if untreated |
| Inheritance Pattern | Autosomal recessive |
Explore related products






What You'll Learn
![]()
Clinical Presentation Differences
21-hydroxylase deficiency, a key enzyme defect in congenital adrenal hyperplasia (CAH), manifests clinically as either the simple-virilizing or salt-wasting form, with distinct presentations that dictate management urgency. The simple-virilizing form typically presents in female infants with ambiguous genitalia at birth, a result of excessive androgen production during fetal development. These infants often have a 46,XX karyotype but exhibit virilized external genitalia, which may include an enlarged clitoris and fused labioscortal folds. In contrast, male infants with this form are usually asymptomatic at birth, as their external genitalia develop normally. The absence of life-threatening electrolyte imbalances in these cases allows for a more elective approach to diagnosis and treatment, often initiated within the first few weeks of life.
The salt-wasting form, however, demands immediate attention due to its severe and potentially fatal clinical presentation. Infants with this variant, regardless of sex, experience profound sodium loss and potassium and hydrogen ion retention, leading to hyponatremia, hyperkalemia, and metabolic acidosis. These electrolyte disturbances can cause poor feeding, vomiting, dehydration, and cardiovascular collapse within the first 2–3 weeks of life. Female infants may also present with ambiguous genitalia, but the primary concern is the critical adrenal insufficiency that requires urgent replacement of glucocorticoids and mineralocorticoids. Without prompt intervention, mortality rates approach 90% within the first month.
A key differentiator in clinical presentation lies in the timing and severity of symptoms. Simple-virilizing CAH is often diagnosed incidentally during routine newborn examinations or when ambiguous genitalia are noted, whereas salt-wasting CAH presents acutely with nonspecific symptoms like poor feeding and lethargy, which can be mistaken for sepsis or other neonatal conditions. Laboratory findings further distinguish the two: salt-wasters exhibit low serum sodium (often <130 mmol/L), elevated potassium (>5.5 mmol/L), and elevated 17-hydroxyprogesterone (17-OHP) levels (>40,000 ng/dL), while simple-virilizers have normal electrolytes and similarly elevated 17-OHP.
Management strategies reflect these clinical differences. For salt-wasting CAH, emergency treatment includes intravenous normal saline to restore intravascular volume, followed by hydrocortisone (50–100 mg/m²/day) and fludrocortisone (0.05–0.2 mg/day) to replace glucocorticoids and mineralocorticoids, respectively. Simple-virilizing CAH allows for a more gradual approach, with hydrocortisone initiated at 12–15 mg/m²/day to suppress androgen production and prevent further virilization. Surgical correction of ambiguous genitalia in females is often deferred until childhood to allow for shared decision-making.
In summary, the clinical presentation of 21-hydroxylase deficiency diverges sharply between simple-virilizing and salt-wasting forms, with the latter posing an immediate threat to life. Recognizing these differences is critical for timely intervention, as it dictates the urgency of treatment and the specific therapeutic approach. Early diagnosis and tailored management are essential to prevent morbidity and mortality in affected infants.
Breathing Out Waste: The Respiratory System's Role in Elimination
You may want to see also
Explore related products






![]()
Hormonal Imbalance Impact
21-hydroxylase deficiency, a key enzyme defect in congenital adrenal hyperplasia (CAH), disrupts cortisol and aldosterone production while allowing androgens to accumulate. This hormonal imbalance manifests in two primary forms: simple-virilizing and salt-wasting. The distinction hinges on aldosterone deficiency, which determines whether the condition threatens life through electrolyte imbalance or primarily affects sexual development. Understanding the hormonal impact is crucial for early diagnosis and tailored management, as both forms share elevated androgen levels but diverge in their systemic consequences.
Analytical Perspective:
In the salt-wasting variant, severe 21-hydroxylase deficiency (residual enzyme activity <1%) leads to profound aldosterone and cortisol deficiency. Without prompt treatment, infants face life-threatening hyponatremia, hyperkalemia, and dehydration within weeks of birth. The hormonal imbalance here is twofold: androgen excess causes prenatal virilization of female fetuses, while aldosterone deficiency collapses electrolyte homeostasis. In contrast, the simple-virilizing form (enzyme activity 1–2%) spares aldosterone production, focusing the impact on androgen-driven virilization and accelerated growth velocity in childhood, with cortisol deficiency managed through chronic glucocorticoid replacement.
Instructive Approach:
For clinicians, distinguishing between these forms requires urgent assessment of serum electrolytes, renin, and androgen levels. Salt-wasting CAH demands immediate mineralocorticoid (e.g., fludrocortisone 0.05–0.2 mg/day in infants) and glucocorticoid (hydrocortisone 10–15 mg/m²/day) therapy to stabilize electrolytes and prevent adrenal crisis. Simple-virilizing cases prioritize glucocorticoid suppression of androgens to normalize growth and prevent precocious puberty, though monitoring for iatrogenic Cushing’s syndrome from overtreatment is essential. Parents should be educated on stress dosing (tripling hydrocortisone during illness) to mitigate adrenal insufficiency risks.
Comparative Insight:
While both forms share a root cause—21-hydroxylase deficiency—their hormonal footprints diverge sharply. Salt-wasting CAH exemplifies the dual crisis of androgen excess and mineralocorticoid failure, requiring dual-hormone replacement. Simple-virilizing CAH, however, isolates androgen excess as the primary concern, with aldosterone production often sufficient to maintain electrolyte balance. This distinction underscores the importance of genotype-phenotype correlation: severe mutations (e.g., *CYP21A2* null alleles) predict salt-wasting, while milder variants may present as simple-virilizing. Genetic testing post-stabilization refines long-term management.
Descriptive Takeaway:
The hormonal imbalance in 21-hydroxylase deficiency paints a spectrum of clinical urgency. In salt-wasting CAH, the absence of aldosterone transforms a metabolic disorder into an acute pediatric emergency, where hours matter. Simple-virilizing CAH, though chronic, carries its own challenges: androgen-driven virilization in females and rapid bone maturation in both sexes threaten fertility and final adult height. Management hinges on balancing hormone replacement to suppress androgens without inducing glucocorticoid toxicity—a delicate task requiring lifelong surveillance and patient-specific titration. Recognizing these hormonal footprints early transforms prognosis, turning a potentially fatal or debilitating condition into a manageable one.
Does Using Find My Friends Drain Your Friend’s Phone Battery?
You may want to see also
Explore related products






![]()
Diagnosis Methods Comparison
21-hydroxylase deficiency, a common cause of congenital adrenal hyperplasia (CAH), presents in two primary forms: simple-virilizing and salt-wasting. Distinguishing between these forms is critical for timely and appropriate management. Diagnosis relies on a combination of clinical presentation, biochemical markers, and genetic testing, each method offering unique advantages and limitations.
Biochemical Testing: The First Line of Inquiry
Initial diagnosis often begins with biochemical assays, particularly measurements of 17-hydroxyprogesterone (17-OHP). Elevated levels of 17-OHP, typically above 200 ng/dL in newborns, are highly suggestive of 21-hydroxylase deficiency. For salt-wasting CAH, additional markers such as low serum sodium (<135 mmol/L), high potassium (>5.5 mmol/L), and elevated plasma renin activity are crucial. Simple-virilizing forms may present with milder electrolyte imbalances or none at all. A key limitation of biochemical testing is its inability to predict disease severity without further investigation, as 17-OHP levels can overlap between the two forms.
Genetic Analysis: Precision in Prediction
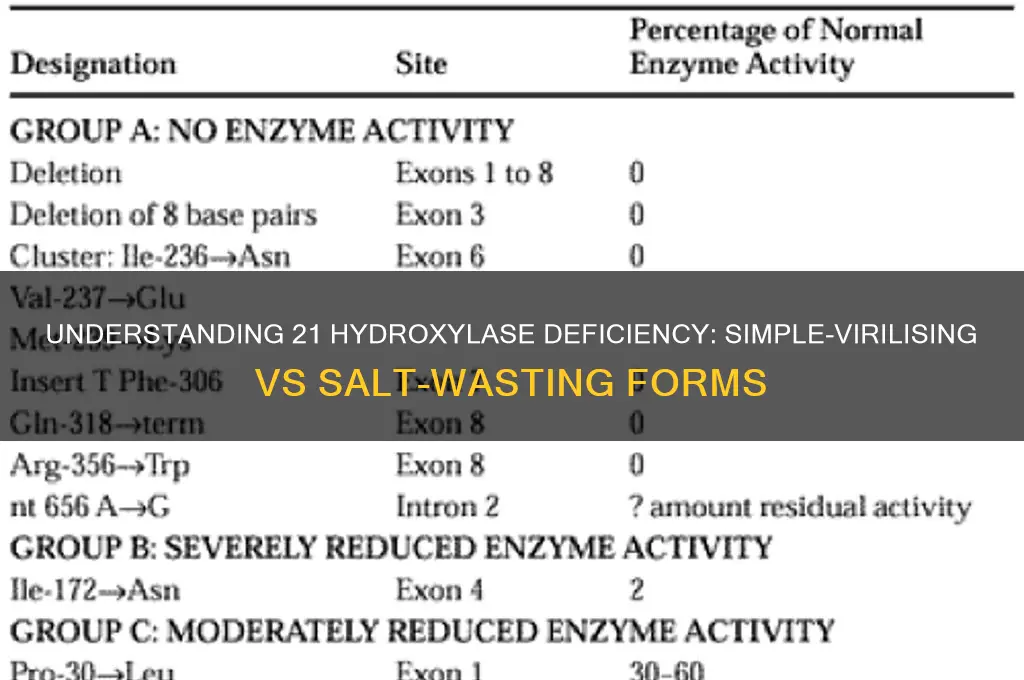
Genetic testing provides a more definitive diagnosis by identifying mutations in the *CYP21A2* gene. Common mutations, such as p.Val281Leu and p.Ile236Met, are associated with varying degrees of enzyme deficiency. Salt-wasting CAH is typically linked to severe mutations resulting in <1% of normal enzyme activity, while simple-virilizing forms often involve milder mutations with 1–2% activity. Genetic analysis not only confirms the diagnosis but also aids in predicting disease severity and guiding long-term management. However, it is time-consuming and may not be immediately available in all settings, making it a secondary tool after biochemical screening.
Clinical Presentation: Contextual Clues
Clinical evaluation remains essential, particularly in distinguishing between the two forms. Salt-wasting CAH presents acutely in neonates with dehydration, hyponatremia, and hyperkalemia, often within the first 2–3 weeks of life. Simple-virilizing CAH, on the other hand, may manifest later with ambiguous genitalia in females or precocious puberty in both sexes, without significant electrolyte disturbances. While clinical signs are informative, they are not definitive and must be corroborated with laboratory findings.
Imaging and Additional Studies: Supporting Evidence
In ambiguous cases, adrenal imaging (e.g., ultrasound or MRI) may reveal enlarged adrenal glands, a common feature in both forms of CAH. However, imaging is not diagnostic and serves primarily to assess adrenal morphology. ACTH stimulation tests, though less commonly used today, can help differentiate between 21-hydroxylase deficiency and other forms of CAH by measuring cortisol and aldosterone responses. These tests, however, are invasive and have largely been replaced by genetic and biochemical methods.
Practical Considerations: Balancing Speed and Accuracy
In emergency settings, such as suspected salt-wasting CAH, rapid biochemical screening for 17-OHP and electrolytes is critical to initiate prompt treatment. Genetic testing, while invaluable for long-term management, is not urgent in these cases. For simple-virilizing CAH, a more comprehensive approach, including genetic analysis and careful clinical monitoring, is warranted. Clinicians must weigh the immediacy of biochemical results against the predictive power of genetic testing to tailor management effectively.
In summary, diagnosing 21-hydroxylase deficiency as simple-virilizing or salt-wasting requires a multifaceted approach. Biochemical testing offers speed and accessibility, genetic analysis provides precision, and clinical evaluation supplies context. Each method complements the others, ensuring accurate diagnosis and appropriate intervention for this complex disorder.
Stop Overspending: Smart Tips to Avoid Wasting Money on Unnecessary Things
You may want to see also
Explore related products






![]()
Treatment Approaches Variation
21-hydroxylase deficiency, a common form of congenital adrenal hyperplasia (CAH), presents in two primary phenotypes: simple-virilizing and salt-wasting. Treatment approaches vary significantly between these forms, driven by the severity of adrenal insufficiency and the presence of androgen excess. The cornerstone of management is glucocorticoid replacement, but the dosage, timing, and additional interventions differ markedly. For instance, salt-wasting CAH requires immediate mineralocorticoid replacement to prevent life-threatening adrenal crises, while simple-virilizing CAH focuses on suppressing androgen production to manage virilization.
In salt-wasting CAH, treatment is urgent and multifaceted. Newborns diagnosed through screening must start hydrocortisone at 10–15 mg/m²/day, divided into two doses, alongside fludrocortisone (0.05–0.2 mg/day) to replace aldosterone. Parents must be educated on stress dosing—doubling or tripling hydrocortisone during illness—to prevent crises. Oral sodium chloride supplementation (2–4 mmol/kg/day) is often necessary until adrenal function stabilizes. Monitoring electrolytes weekly initially, then monthly, ensures adequacy of mineralocorticoid replacement. This aggressive approach contrasts with simple-virilizing CAH, where mineralocorticoid replacement is rarely needed.
For simple-virilizing CAH, the focus shifts to glucocorticoid suppression of androgen synthesis while minimizing growth suppression and iatrogenic Cushing’s syndrome. Hydrocortisone doses range from 8–12 mg/m²/day, often in three divided doses to mimic physiological rhythms. Some clinicians prefer prednisone (0.5–1 mg/kg/day) or dexamethasone (0.05–0.15 mg/kg/day) for their longer half-lives, but these carry higher risks of side effects. Regular monitoring of bone age, growth velocity, and androgen levels guides dose adjustments. Unlike salt-wasting CAH, simple-virilizing cases require long-term management of genital virilization and fertility concerns, often involving surgical consultation for ambiguous genitalia.
A critical comparison reveals that while both forms require lifelong glucocorticoid therapy, the salt-wasting variant demands immediate, high-dose mineralocorticoid replacement and electrolyte monitoring, whereas simple-virilizing CAH prioritizes androgen control and growth preservation. The takeaway is that treatment must be tailored to the phenotype, balancing hormone replacement with the risks of overtreatment. For example, over-suppression in simple-virilizing CAH can stunt growth, while under-replacement in salt-wasting CAH risks dehydration and shock. Clinicians must individualize therapy, considering age, pubertal status, and patient-specific goals.
Practical tips include using wearable medical alert bracelets for salt-wasting patients to ensure emergency responders recognize adrenal crisis risk. For simple-virilizing CAH, engaging psychologists early can help adolescents navigate body image and gender identity concerns. Parents should be taught to recognize signs of adrenal insufficiency (e.g., vomiting, lethargy) and keep emergency hydrocortisone injections accessible. Annual multidisciplinary team reviews—including endocrinologists, surgeons, and psychologists—ensure holistic care. Ultimately, treatment variation reflects the distinct challenges of each phenotype, requiring precision and adaptability.
Giant Tube Worms' Unique Waste Disposal Methods Explained
You may want to see also
Explore related products






![]()
Long-Term Outcomes Analysis
21-hydroxylase deficiency, a hallmark of congenital adrenal hyperplasia (CAH), manifests in two primary phenotypes: simple-virilizing and salt-wasting. Long-term outcomes analysis reveals distinct trajectories for each, shaped by early intervention, treatment adherence, and the inherent severity of the condition. For salt-wasting CAH, prompt diagnosis and initiation of glucocorticoid and mineralocorticoid replacement within the first week of life are critical. Delayed treatment increases the risk of adrenal crises, which, over time, can lead to hypertension, growth retardation, and cognitive impairments. In contrast, simple-virilizing CAH, often diagnosed later due to less acute symptoms, presents challenges related to androgen excess, including premature pubarche, accelerated bone aging, and psychosocial distress.
Analyzing growth outcomes, children with salt-wasting CAH often require higher initial hydrocortisone doses (12-18 mg/m²/day) to prevent crises, but long-term overtreatment can suppress growth. Regular monitoring of bone age and height velocity is essential, with dose adjustments every 3-6 months to balance adrenal insufficiency and iatrogenic Cushing’s syndrome. For simple-virilizing CAH, the focus shifts to managing androgen-driven complications. Girls may require surgical intervention for ambiguous genitalia, followed by lifelong psychological support to address body image and gender identity concerns. Boys, though less affected, may experience early androgen exposure, necessitating careful monitoring of bone maturation and height potential.
A comparative analysis highlights the role of adherence in long-term outcomes. Adolescents with both phenotypes face challenges in treatment compliance, particularly during puberty, when glucocorticoid requirements may fluctuate. Poor adherence in salt-wasting CAH increases the risk of recurrent crises, while in simple-virilizing CAH, it exacerbates androgen-related complications. Practical strategies include transitioning to once-daily hydrocortisone formulations (e.g., 10-15 mg/m² in the morning) and incorporating stress dosing education for illness or injury. Digital health tools, such as medication reminders and telehealth follow-ups, have shown promise in improving adherence among teens.
Persuasively, fertility and reproductive health emerge as critical long-term considerations. Women with simple-virilizing CAH often face polycystic ovary syndrome (PCOS)-like symptoms, requiring tailored management to optimize fertility. Men, though less studied, may experience subfertility due to androgen excess. For both phenotypes, preconception counseling is vital, emphasizing the need for stable hormone levels before pregnancy to minimize fetal virilization risks. During pregnancy, hydrocortisone doses may need to triple, necessitating close endocrinological oversight.
Descriptively, the psychosocial landscape of 21-hydroxylase deficiency is complex. Children with ambiguous genitalia or early pubertal changes may face bullying or stigma, underscoring the need for integrated mental health support. Schools and caregivers should be educated on CAH to foster inclusivity. Adult survivors often report anxiety, depression, and body dissatisfaction, particularly in simple-virilizing CAH. Support groups and access to specialized care teams can mitigate these challenges, fostering resilience and quality of life. In conclusion, long-term outcomes in 21-hydroxylase deficiency hinge on individualized, multidisciplinary care, with a focus on preventing complications, optimizing adherence, and addressing holistic well-being.
Universities and E-Waste: Exploring the Role of Recyclers in Education
You may want to see also
Frequently asked questions
21 hydroxylase deficiency is a genetic disorder that affects the adrenal glands' ability to produce cortisol and aldosterone, leading to a range of symptoms depending on the severity of the deficiency.
21 hydroxylase deficiency can present in two main forms: simple-virilizing and salt-wasting. The salt-wasting form is more severe and characterized by low levels of aldosterone, leading to salt and water loss, while the simple-virilizing form primarily affects cortisol production and can cause virilization in females.
Symptoms of simple-virilizing 21 hydroxylase deficiency include ambiguous genitalia in females, early appearance of pubic hair, and rapid growth in childhood, followed by early cessation of growth and short stature in adulthood.
Symptoms of salt-wasting 21 hydroxylase deficiency include vomiting, dehydration, low blood pressure, high potassium levels, and low sodium levels, often presenting in infancy or early childhood, and can be life-threatening if left untreated.